Page 72 - Perdih, Andrej, Katja Lakota, Alja Prah. 2020. Strukture bioloških molekul. Univerzitetni učbenik z recenzijo in navodila za vaje. Koper: Založba Univerze na Primorskem.
P. 72
andrej perdih, katja lakota, alja prah
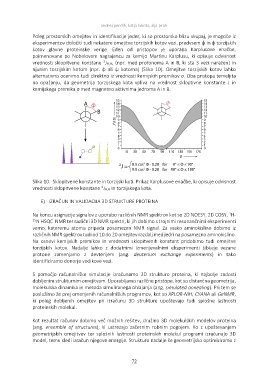
Poleg prostorskih omejitev in identifikacije jeder, ki so prostorsko blizu skupaj, je mogoče iz
eksperimentov določiti tudi nekatere omejitve torzijskih kotov vezi, predvsem ψ in ϕ torzijskih
kotov glavne proteinske verige. Eden od pristopov je uporaba Karplusove enačbe,
poimenovane po Nobelovem nagrajencu za kemijo Martinu Karplusu, ki opisuje odvisnost
vrednosti sklopitvene konstane 3JH,H, (npr. med protonoma A in B, ki sta 3 vezi narazen) in
njunim torzijskim kotom (npr. ϕ ali ψ kotoma) (Slika 10). Omejitve torzijskih kotov lahko
alternativno ocenimo tudi direktno iz vrednosti kemijskih premikov σ. Oba pristopa temeljita
na opažanju, da geometrija torzijskega kota vpliva na vrednost skloptivne konstante J in
kemijskega premika σ med magnetno aktivnima jedroma A in B.
Slika 10. Sklopitvene konstante in torzijski koti. Prikaz Karplusove enačbe, ki opisuje odvisnost
vrednosti sklopitvene konstane 3JH,H in torzijskega kota.
E) IZRAČUN IN VALIDACIJA 3D STRUKTURE PROTEINA
Na koncu asignacije signalov z uporabo različnih NMR spektrov kot so 2D NOESY, 2D COSY, 1H-
15N HSQC NMR ter različni 3D NMR spektri, ki jih dobimo s trojnimi resonančnimi eksperimenti
vemo, kateremu atomu pripada posamezen NMR signal. Za vsako aminokislino dobimo iz
različnih NMR spektrov tudi od 10 do 20 omejitev razdalj med jedri na posamezno aminokislino.
Na osnovi kemijskih premikov in vrednosti sklopitvenih konstant pridobimo tudi omejitve
torzijskih kotov. Nadalje lahko z dodatnimi izmenjevalnimi eksperimenti šibkeje vezane
protone zamenjamo z devterijem (ang. deuterium exchange experiments) in tako
identificiramo donorje vodikove vezi.
S pomočjo računalniške simulacije izračunamo 3D strukturo proteina, ki najbolje zadosti
dobljenim strukturnim omejitvam. Uporabljamo različne pristope, kot so distančna geometrija,
molekulska dinamika in metoda simuliranega ohlajanja (ang. simulated annealing). Pri tem se
poslužimo že prej omenjenih računalniških programov, kot so XPLOR-NIH, CYANA ali GeNMR,
ki poleg dobljenih omejitev pri izračunu 3D strukture upoštevajo tudi splošne lastnosti
proteinskih molekul.
Kot rezultat računov dobimo več možnih rešitev, družino 3D molekulskih modelov proteina
(ang. ensemble of structures), ki ustrezajo začetnim robnim pogojem. Ko z upoštevanjem
geometrijskih omejitvev ter splošnih lastnosti proteinskih molekul programi izračunajo 3D
model, temu sledi izračun njegove energije. Strukturo nadalje še geometrijsko optimiziramo z
72
Poleg prostorskih omejitev in identifikacije jeder, ki so prostorsko blizu skupaj, je mogoče iz
eksperimentov določiti tudi nekatere omejitve torzijskih kotov vezi, predvsem ψ in ϕ torzijskih
kotov glavne proteinske verige. Eden od pristopov je uporaba Karplusove enačbe,
poimenovane po Nobelovem nagrajencu za kemijo Martinu Karplusu, ki opisuje odvisnost
vrednosti sklopitvene konstane 3JH,H, (npr. med protonoma A in B, ki sta 3 vezi narazen) in
njunim torzijskim kotom (npr. ϕ ali ψ kotoma) (Slika 10). Omejitve torzijskih kotov lahko
alternativno ocenimo tudi direktno iz vrednosti kemijskih premikov σ. Oba pristopa temeljita
na opažanju, da geometrija torzijskega kota vpliva na vrednost skloptivne konstante J in
kemijskega premika σ med magnetno aktivnima jedroma A in B.
Slika 10. Sklopitvene konstante in torzijski koti. Prikaz Karplusove enačbe, ki opisuje odvisnost
vrednosti sklopitvene konstane 3JH,H in torzijskega kota.
E) IZRAČUN IN VALIDACIJA 3D STRUKTURE PROTEINA
Na koncu asignacije signalov z uporabo različnih NMR spektrov kot so 2D NOESY, 2D COSY, 1H-
15N HSQC NMR ter različni 3D NMR spektri, ki jih dobimo s trojnimi resonančnimi eksperimenti
vemo, kateremu atomu pripada posamezen NMR signal. Za vsako aminokislino dobimo iz
različnih NMR spektrov tudi od 10 do 20 omejitev razdalj med jedri na posamezno aminokislino.
Na osnovi kemijskih premikov in vrednosti sklopitvenih konstant pridobimo tudi omejitve
torzijskih kotov. Nadalje lahko z dodatnimi izmenjevalnimi eksperimenti šibkeje vezane
protone zamenjamo z devterijem (ang. deuterium exchange experiments) in tako
identificiramo donorje vodikove vezi.
S pomočjo računalniške simulacije izračunamo 3D strukturo proteina, ki najbolje zadosti
dobljenim strukturnim omejitvam. Uporabljamo različne pristope, kot so distančna geometrija,
molekulska dinamika in metoda simuliranega ohlajanja (ang. simulated annealing). Pri tem se
poslužimo že prej omenjenih računalniških programov, kot so XPLOR-NIH, CYANA ali GeNMR,
ki poleg dobljenih omejitev pri izračunu 3D strukture upoštevajo tudi splošne lastnosti
proteinskih molekul.
Kot rezultat računov dobimo več možnih rešitev, družino 3D molekulskih modelov proteina
(ang. ensemble of structures), ki ustrezajo začetnim robnim pogojem. Ko z upoštevanjem
geometrijskih omejitvev ter splošnih lastnosti proteinskih molekul programi izračunajo 3D
model, temu sledi izračun njegove energije. Strukturo nadalje še geometrijsko optimiziramo z
72